Martin Lab
The Martin Lab is dedicated to the development of novel gene therapy treatments for patients with a variety of neurological, neuromuscular and lysosomal storage disorders. The lab has pioneered dual gene AAV therapies to both replace lost gene function and rebuild lost muscle mass and strength in forms of muscular dystrophy and myopathy, including Duchenne muscular dystrophy (DMD), Limb-girdle muscular dystrophy and GNE myopathy. It is predicted that this approach will allow for new, more potent therapies to be developed for at least 23 forms of human muscle disease. At least one of these therapies may also be applied to the loss of muscle mass during normal aging, termed sarcopenia. The lab has also developed a liver-directed therapy for lysosomal acid lipase deficiency (LALD) and is working on a gene therapy to treat another lipid disease, Neutral Lipid Storage Disease with Myopathy (NLSD-M). These therapies may also have impacts in other diseases where lipid imbalances occur, such as coronary artery disease, non-alcoholic fatty liver disease and non-alcoholic steatohepatitis.
Inside the Martin Lab
Our Research
There is a profound need to improve gene therapy treatments for patients with muscle diseases, particularly patients where the disease is already present and has caused a loss of muscle mass and strength. The Martin Lab is engineering multi-functional single-treatment AAV therapies with the goal of allowing such patients to recover muscle strength and regain more normal muscle function.
The lab tests novel therapies in a variety of genetic models of disease. We engineer AAV gene therapies that have optimized gene replacement therapy to stop the progression of disease and coupled this with a muscle-building therapy to rebuild lost muscle mass and strength. Once we have optimized such therapies and defined the doses that give rise to optimal impacts on disease, we then test the safety of these vectors with the goal of moving forward with phase 1/2a first in human clinical trials to determine if these therapies will benefit patients.
Featured Publications
- Liver-directed AAV gene therapy normalizes disease symptoms and provides cross-correction in a model of lysosomal acid lipase deficiency
- Dual FKRP/FST gene therapy normalizes ambulation, increases strength, decreases pathology, and amplifies gene expression in LGMDR9 mice
- Development of Assays to Measure GNE Gene Potency and Gene Replacement in Skeletal Muscle
- A first-in-human phase I/IIa gene transfer clinical trial for Duchenne muscular dystrophy using rAAVrh74.MCK. GALGT2
- Micro-laminin gene therapy can function as an inhibitor of muscle disease in the dyW mouse model of MDC1A
Join Our Team
The Martin Lab is always growing. If you are interested in collaborating or joining our team as a postdoctoral scientist, please email us at Paul.Martin@NationwideChildrens.org.
Featured Research Projects
Neutral lipid storage disease with myopathy (NLSD-M) is an ultrarare autosomal recessive disease caused by mutations in the PNPLA2 gene, which encodes the enzyme adipose triglyceride lipase (ATGL). The PNPLA2 gene is required for the cleavage of triglycerides within lipid droplets to free fatty acids. Patients with NLSD-M have a deficiency in triglyceride cleavage and a build-up of triglycerides and lipid droplets in tissues throughout the body. This lipid build-up greatly affects skeletal muscles, causing myopathy and weakness. Additional pathologies are also present in liver (fatty liver disease) and heart (atherosclerosis and cardiomyopathy). The goal of this research project in the Martin lab is to develop and test a potentially curative gene therapy treatment for patients with NLSD-M, and then to translate that gene therapy to the clinic for patients.
Research will first involve optimizing a human gene therapy to treat NLSD-M by testing various promoters, enhancers, and AAV serotypes to identify a medicine that can be used safely and effectively at a very low dose. Proof of concept and safety studies will be done on this human gene therapy with the goal of translating this therapy to the clinic for NLSD-M patients. Tests of the efficacy of gene therapy to reverse lipid build-up and disease symptoms will be done in cells from patients and in a mouse model of the disease.
The first year of this project has been funded by a generous gift from the Reisenauer Precision Medicine Fund to the Center for Gene Therapy at Nationwide Children’s Hospital.
.jpg)
The power of dual gene therapy. Figure shows staining of muscle cells for functionally glycosylated alpha dystroglycan. LGMDR9 is caused by mutations that reduce functional glycosylation of alpha dystroglycan. Here, orange-red shows very high glycosylation, yellow-green shows intermediate levels of glycosylation, and blue-magenta shows very low levels of glycosylation. Upper left panel shows a wild type (normal) muscle. Upper right panel shows a muscle from an FKRP mutant model of LGMDR9. Bottom right panel shows an FKRP mutant treated with FKRP gene therapy (gene replacement), bottom left panel shows an FKRP mutant treated with FST gene therapy (muscle building), center panel shows an FKRP mutant treated with a dual FKRP/FST gene therapy. The dual therapy not only recovers glycosylation beyond normal levels, but it dramatically increases the size muscle fibers, also increasing total muscle mass and strength. Dual gene therapy can recover normal (wild type) levels of ambulation in the FKRP mutant and can build muscle mass beyond normal levels.
This project involves the development of a first-in-class dual AAV gene therapy for the treatment of patients with Limb Girdle Muscular Dystrophy Type R9, which arises from mutations in the FKRP gene. The project will engineer new therapies to optimize the rebuilding of lost muscle mass and strength in patients who already have significant disease.
Thank you to National Institutes of Health for funding this project.
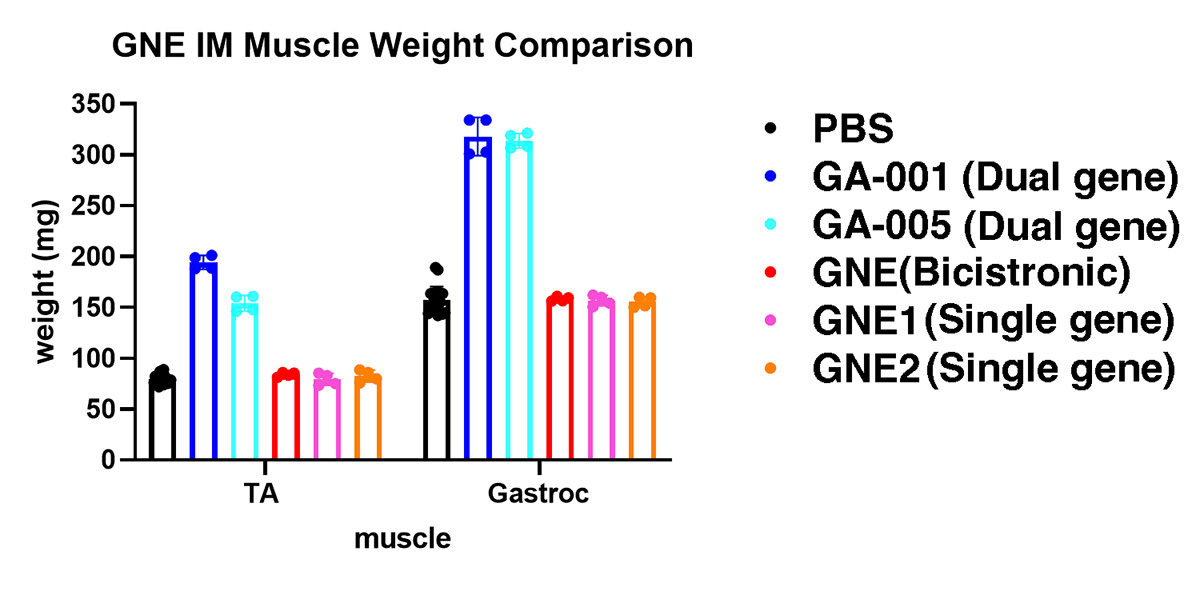
Weights of the tibialis anterior muscle (TA) and gastrocnemius muscle (Gastroc) after a 2 month treatment with various GNE gene therapy vectors. GA-001 and GA-005 are dual gene therapy candidates that provide both gene replacement, to prevent subsequent disease, and a muscle building therapy, to build new muscle mass and strength. Both the TA and Gastroc muscle mass doubles in size in only two months after dual gene therapy treatment. A bicistronic IRES construct, which is build in a different way than our dual gene therapy technologies, did not build new muscle mass, and several single GNE gene replacement AAV vectors, which lack a muscle building gene, also built no new muscle mass. These dual gene therapy vectors provide hope for patients with pre-existing disease, as they can build back stronger muscles in patients where muscle weakness is already present.
The goal of this project is to design a first-in-class dual AAV gene therapy for the treatment of GNE myopathy. Patients with GNE myopathy have muscle disease as the result of recessive loss of function mutations in the GNE gene, a gene that encodes a dual enzyme responsible for the committed steps in sialic acid biosynthesis. The lab is developing a variety of models for loss of GNE gene function that speak to the various diseases caused by mutations in the GNE gene, including infantile thrombocytopenia, juvenile amyotrophic lateral sclerosis (ALS), and GNE myopathy, which occurs in teenagers and adults.
The lab is building dual gene AAV gene therapies to replace missing GNE gene function (gene replacement), which will prevent subsequent disease, and coupling this with a muscle building therapy to grow new muscle, building new muscle mass and strength. Such an approach is needed to treat patients who already have significant disease at the time of diagnosis, which is almost all patients with GNE myopathy.
Thank you to the Neuromuscular Disease Foundation, Solve GNE and the Technology Development Fund at Nationwide Children's Hospital for funding this project.
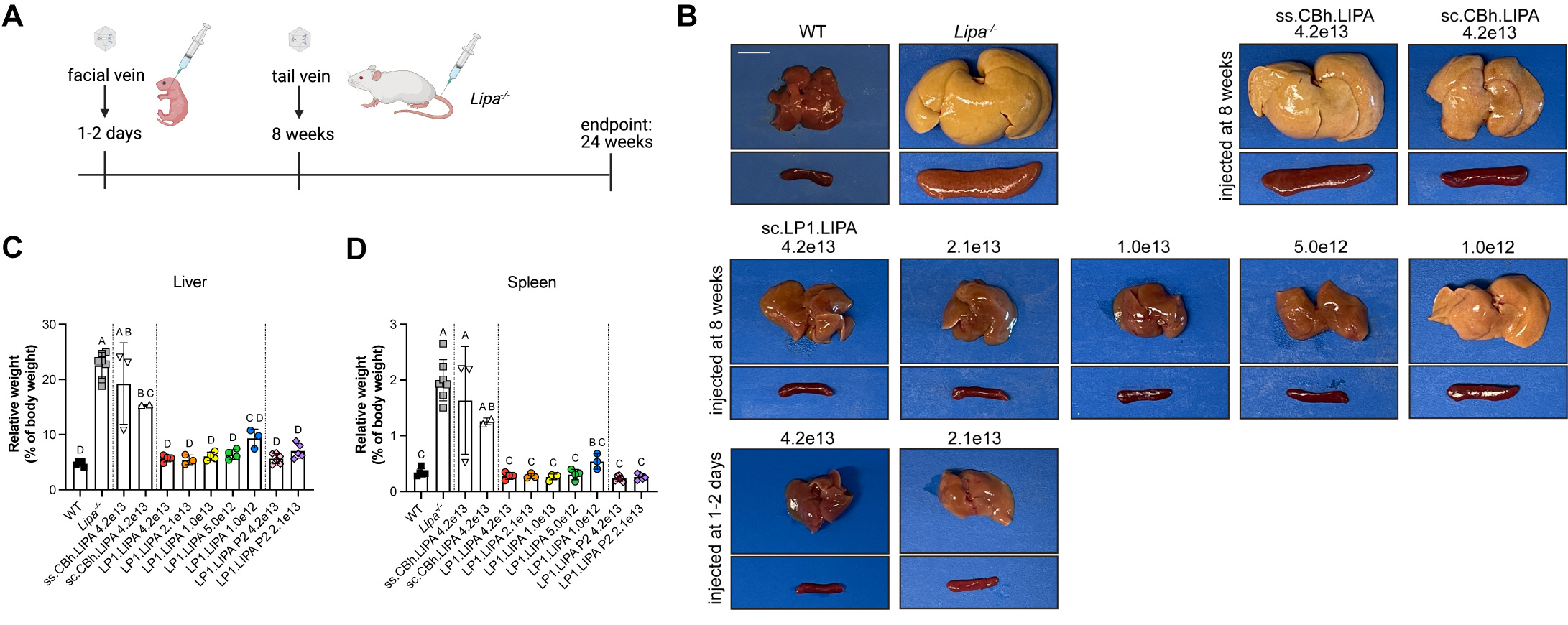
(A) An LAL-D model (Lipa-/-) was injected intravenously at postnatal day 1-2, prior to disease, or at 8 weeks, when disease was already very significant. (B) Examples of livers (above) and spleens (below) show large increases in weight in LAL-D due to fat deposition, which also discolors the organs. Liver directed therapy (LP1) shows clear dose-depending reversal in liver (C) and spleen (D) weight, while promoters that express in all organs (Cbh) fail to do so due to induction of immune responses to the treatment.
The goal of this project is to develop a single treatment curative therapy for Lysosomal Acid Lipase Deficiency, or LAL-D. LAL-D, is a lysosomal storage disorder that develops in patients with recessive, loss of function, mutations in the Lipase A (LIPA) gene. Lipase A is the only lysosomal enzyme that breaks down triglycerides to free fatty acids and cholesteryl esters into free cholesterol. Absence of LIPA function leads to severe lipid imbalances in patients, with increased triglyceride and cholesterol deposition in organs, particularly the liver.
We have built a liver-directed AAV gene therapy to increase LIPA enzyme activity in the liver that also secretes this enzyme into the blood, delivering it to other tissues, where it is taken up and transported to the lysosome to replace lost enzyme activity (a phenomenon called cross-correction). We have shown that this gene therapy not only can prevent LAL-D from occurring but that it can reverse disease symptoms that are already present. While not yet proven, this gene therapy may also be applied to diseases caused by obesity, including coronary artery disease, non-alcoholic fatty liver disease, and non-alcoholic steatohepatitis.
Thank you to LAL-D Aware, AE LALD and the Technology Development Fund at Nationwide Children's Hospital for funding this project.
The Martin Lab is collaborating with the INADCure Foundation to provide toxicology studies to test the safety of PLA2G6 gene therapy for use in patients with Infantile Neuroaxonal Dystrophy, or INAD.
Thank you to the INADCure Foundation for funding this project.


