Bradbury Lab
The Bradbury Laboratory is dedicated to understanding disease mechanisms and developing therapeutic approaches for rare, pediatric neurodegenerative disorders. Advances in adeno-associated virus (AAV) vectors have led to safer and more efficient viral vehicles to deliver therapeutic transgenes in a single injection, and gene therapy is now a favorable therapeutic intervention for monogenic diseases. A primary focus of the Bradbury Lab is improving AAV targeting of myelinating cells, a current limitation of AAV, in order to advance therapeutic outcomes for leukodystrophies and other white matter disorders. The Bradbury Lab is translational in nature with a commitment to safely and efficiently moving therapies into the clinic for rare, pediatric disorders.
Inside Bradbury Lab
Our Research
The goal of our research is to translate safe and effective genetic medicines from bench to bedside for devastating pediatric neurological disorders, many of which currently have no therapies available.
Our team utilizes a wide variety of techniques to most effectively answer questions about disease mechanisms and therapeutic efficacy. We leverage patient-derived cell lines, organoid model systems, and bespoke mouse models, to name a few. We also explore a wide variety of therapeutic strategies including AAV-mediated gene replacement, antisense oligonucleotides, gene editing and small molecule drugs.
If you are interested in collaborating with the Bradbury Lab, please email Nettie Pyne at Nettie.Pyne@NationwideChildrens.org.
Bradbury Lab News Articles
Personalized Therapy for SLC6A1 Disorder
- The Texas Mom Who Custom-Ordered Mice to Save Her Son
- Setting the Stage the Next Era of Gene Therapy for Ultrarare Disease
- Nationwide Children's gene therapy shows early promise after Taysha pull-back
- 8-Year-Old Has Disease So Rare It Doesn't Have a Name. Now He's Frist in the World to Receive Treatment
- Let's drop the Midwest modesty. Columbus changing world, lives | Opinion
Therapy Development for TBCD-related Encephalopathy
Therapy Development for Vanishing White Matter Disease
A Novel Gene Correction REMEDY for Genetic Disorders
Feature on Dr. Allison Bradbury
Research Projects

The goal of the project is to characterize, optimize, and validate AAV-mediated EIF2B5 gene replacement therapy, including evaluation of a novel astrocyte-targeting promoter, with the ultimate goal to prevent or mitigate VWM disease.
Work at Nationwide Children's Hospital has included characterizing a VWM mouse model, as well as extensive testing and optimization of AAV-mediated therapies, to determine which therapy and dose is the most effective at mitigating VWM disease in the mice.
We have also begun work on a combination therapy using both our AAV based therapy, in addition to the small molecule drug ISRIB, that is currently in clinical trials for VWM.
This work is funded by a Sponsored Research Agreement from the Columbus Foundation, and the combination therapy work is funded by an NIH F99 (PI Herstine, 1F99NS139538) and a grant from ELA International.
The major goals of this project are to develop a targeted recombinant AAV gene therapy approach for SLC6A1 related neurodevelopmental disorder.
We have developed and performed preclinical testing on a number of iterations of targeted AAV-mediated therapy, determined a lead candidate, performed an investigational new drug (IND) enabling toxicology study and received IND approval in 2025. This gene therapy is now in an open clinical trial for a specific variant. Additional preclinical studies are underway to determine if the clinical trial can be expanded.
- Setting the Stage the Next Era of Gene Therapy for Ultrarare Disease
- Nationwide Children's Gene Therapy Shows Early Promise After Taysha Pull-Back
We are now performing further testing to support expanded use of this therapeutic in more patients.
This work is funded by the SLC6A1 Connect foundation, and was previously funded by the Simons Foundation Autism Research Initiative (SFARI).
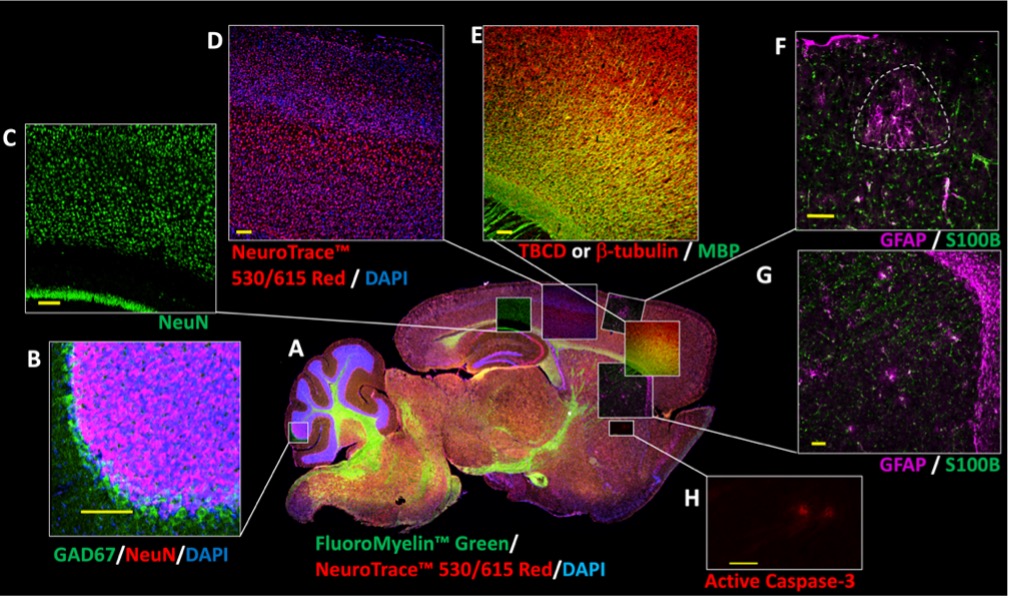
Biallelic mutations in the tubulin folding cofactor D (TBCD) gene result in a severe early-onset developmental and epileptic encephalopathy for which there is no treatment. We are working to determine the neuropathological mechanisms of TBCD variants using patient-derived cerebral organoids, and are investigating the molecular and clinical phenotypes in a corresponding allelic series of TBCD knock-in mouse models. We will then evaluate the efficacy of a targeted AAV gene replacement therapy in established models.
We have made great strides in collecting patient genomic data and samples, and in generating cerebral organoids for study. We are also actively working on characterizing multiple candidate mouse models to determine which will be the most useful for therapy testing.
This work is funded by an NIH R01 Grant (R01NS134923), and preliminary work was funded by generous gifts from The TBCD Foundation.

The major goal of this project is to attempt an in-utero gene therapy for Krabbe disease in the canine model system.
Dr. Bradbury's previously published works demonstrated that AAV-mediated gene therapy was effective at mitigating disease in the canine model when administered at a very early time point (2 weeks of age). However, there was still some evidence of plaques in the brain despite the dramatic extension in lifespan, indicating that some damage to the brain had already occurred by 2 weeks of age. Further, it has long been known that the cytotoxic substrate, psychosine, is detectable during the fetal stage in human patients. This implies that even earlier treatment would be beneficial. In collaboration with Cincinnati Children's Hospital and the Auburn College of Veterinary Medicine, we are attempting to deliver the AAV therapy to diseased animals in utero, to establish if more complete prevention of the disease is possible.
This work is funded by the Rosenau Family Research Foundation and Cincinnati Children's Hospital Medical Center, North American Fetal Therapy Network (NAFTNET).

There are two major goals in this project:
- To develop an N of 1 antisense oligonucleotide therapy for a specific splice site variant.
- To develop a dual-vector AAV-mediated gene replacement therapy that would work for most or all patients with ATRX syndrome.
For the ASO therapy, we are beginning a toxicology study on our lead candidate ASO as a necessary step to translating this precision therapy to the clinic.
As ATRX is too large to fit in a single AAV, for a universal therapy, we are pursing a dual-vector AAV-mediated gene replacement. We are also working on characterizing a new mouse model of ATRX-syndrome and patient-derived organoid models for future therapeutic testing.
The ASO toxicology study is funded by the Reisenauer Precision Medicine Fund (030-92421-993) and matching gift funds.
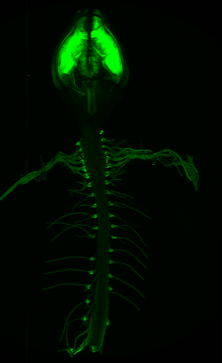
The major goal of this project is to develop a Schwann cell targeted recombinant AAV gene therapy approach for the treatment of NF1.
In collaboration with UMass Chan Medical School, we are utilizing AAV-mediated gene replacement of a NF1 mini gene developed by Miguel Sena-Esteves. We are currently evaluating expression of this mini gene in the context of different promoters and delivery routes to determine optimal targeting of Schwann cells.
This work is funded by the Gilbert Family Foundation (521003).
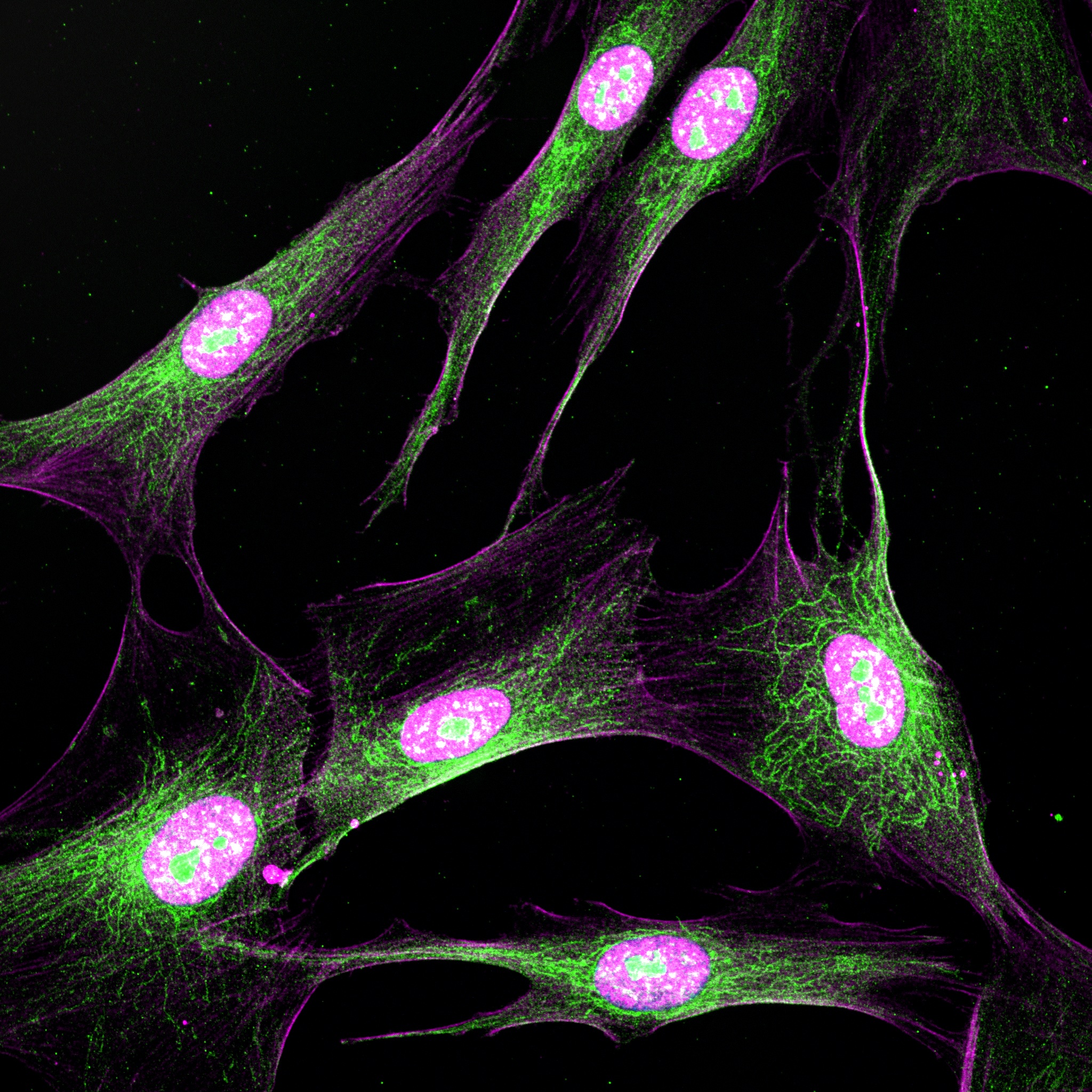
The goal of this study is to characterize and expand research on a novel patient-derived NF2 induced Schwann cell model to determine its full potential for broader NF2 disease mechanism studies, and for testing novel therapeutics in a patient-specific manner.
We are working to characterize a novel, in vitro, patient-specific model system for NF2 involving converting patient fibroblasts (skin cells) into induced Schwann cells, which will allow us to both better understand the mechanisms of disease in NF2, as well as test therapeutics in a patient specific manner. We are working on testing both AAV-mediated and drug therapies in this system.
The project is funded by the Department of Defense (HT9425-24-1-0616).


