Extracellular Vesicles in Monitoring and Treatment of Dystrophinopathy
Principal Investigator

Nizar Saad, PhD
Dr. Nizar Y. Saad is a is a principal investigator in the Jerry R. Mendell Center for Gene Therapy at Nationwide Children’s Hospital, and an assistant professor at The Ohio State University College of Medicine. He serves as associate director of the Paul D. Wellstone Muscular Dystrophy Specialized Research Center at Nationwide Children’s Hospital.
What Is the Goal of the Project?
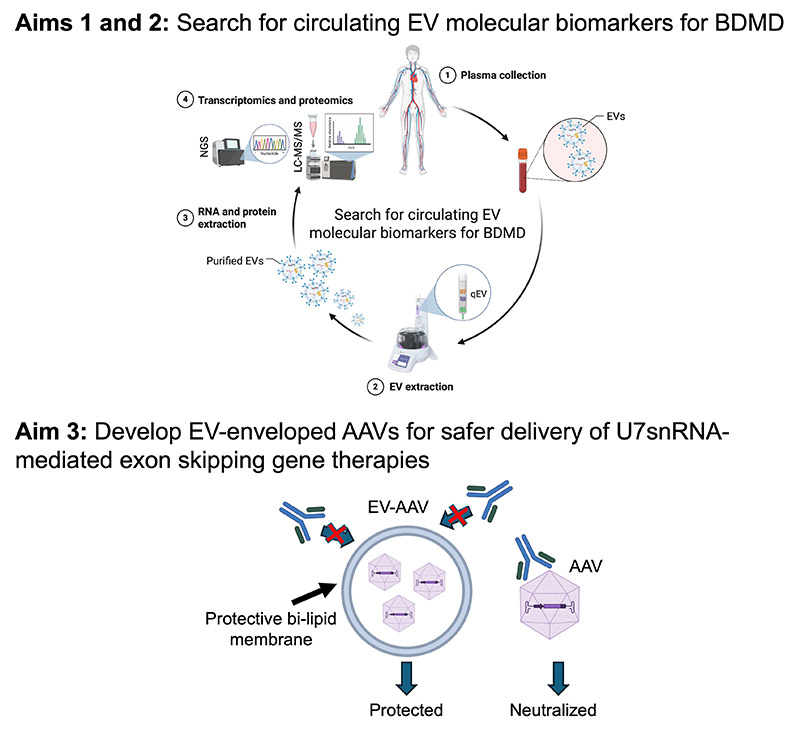
Research Project 3 of our MDSRC Program is aimed to identify DMD circulating EV biomarkers and develop EV-enveloped AAV gene therapy for DMD to shield AAV from anti-AAV antibodies and to increase its transduction efficiency and therapeutic potency.
What Does This Project Entail?
X-linked recessive Duchenne Muscular Dystrophy (DMD) is the most common muscular dystrophy affecting 1 in 3,500 boys. DMD is caused by mutations in the DMD gene, preventing the production of a fully functional dystrophin protein, which normally performs a critical structural role in stabilizing myofiber membranes during muscle contraction. Functional and clinical outcome measures for DMD were used in the clinical trial that led to the recent FDA-approval of the microdystrophin gene replacement therapy. The U7snRNA-based exon skipping is another promising one time single-dose gene therapy, aiming to restore full-length dystrophin expression, that entered clinical trial. The latter relies on similar outcome measures to validate therapeutic efficacy in treated patients. These outcome measures can be labor intensive, time consuming, inconvenient for some patients and expensive, which might delay approval of DMD therapies. Moreover, it is still unknown how efficacious these gene therapies would be in the larger DMD population and for how long their therapeutic benefits will last. Therefore, non-invasive, longitudinal monitoring of treated patients is needed to understand disease progression and assess persistence of therapeutic interventions and safer next-generation gene therapies with higher potency and possibility of re-administration are needed. In this project, we will rely on extracellular vesicles (EVs) to meet these needs. To identify novel circulating DMD and BMD biomarkers, we will investigate the RNA and protein content of circulating EVs collected from the plasma of DMD and BMD patients and healthy controls. EVs are lipid-bilayer delimited vesicles released by tissues or organs into biofluids and carry tissue/organ-specific molecules indicative of the pathophysiological state of their tissue or organ of origin. These molecules can be actively enriched in EVs. Therefore, their identification might allow the validation of EV-associated biomarkers that help track DMD and BMD onset and progression, stratify patients for trials and serve as therapy responsive outcomes (Aim 1 and 2). AAV-mediated gene therapy for muscle diseases typically requires systemic delivery, which is challenging for first-generation AAV vectors due to high dose requirements for widespread muscle transduction. High dose, vascular AAV delivery has been linked to adverse events such as liver toxicity and AAV immunogenicity, which can reduce therapeutic efficacy and prevent administration to patients with pre-existing immunity to AAV. To overcome the challenges associated with systemic delivery of high AAV doses, we will package AAV with EVs. By enveloping AAVs with a lipid-bilayer, EVs may reduce total AAV capsid exposure, which would reduce the development of humoral immunity and shield AAV capsids from pre-existing anti-AAV antibodies. Previous work on EV-AAVs have demonstrated that enveloped AAVs were protected from anti-AAV neutralization. We will test the transduction efficiency and therapeutic potency of EV-AAVs in wild-type and DMD mice using as proof-of-concept AAV gene therapies investigated in Project 2 (Aim 3).
